
Annex 19 Has Been Rewritten. If You Touch Parallel Trade, Read Section 9 Before September.
A Just Ask breakdown of the 2026 revision to EU GMP Annex 19: Reference and Retention Samples
The European Commission has issued a revised Annex 19 under Commission Decision C(2026) 4135 (dated 23 June 2026). It applies from 24 September 2026, three months after publication, and replaces the 2006 text that has been in force since 1 June 2006.
The headline for most sites: this is not a big deal. If you don't repackage parallel-traded product, the core reference-and-retention regime you already run is untouched. Sections 1 to 8 and 10 are substantively the same. Your storage durations, sample sizes, storage conditions, written agreements and closedown obligations all survive intact.
The change is almost entirely in Section 9 (parallel imported, distributed and traded products), and it is a real one. The old two-paragraph, risk-based approach has been replaced with a prescriptive set of positive obligations, plus a new digital-sample option. If you are an MAH, QP, importer, re-packager or batch-release site in the parallel-trade chain, this needs a controlled SOP review before the application date.
Talk to us
Where do you start? A gap assessment against the new Section 9 is the fastest way to find out whether your SOPs already comply or need updating, before September arrives.
Below: what actually changed, quote for quote; then ten questions for your impact assessment; what to look at in change control; and four practical options.
The timeline at a glance: 1 Jun 2006 original Annex 19 in force → 17 Oct 2025 veterinary GMP implementing regulations adopted → 23 Jun 2026 revised Annex 19 adopted (C(2026) 4135) → 16 Jul 2026 veterinary GMP regulations apply → 24 Sep 2026 revised Annex 19 applies (your hard deadline).
Part 1: The Differential — Old vs New, Side by Side
The change that matters: Section 9
The 2006 text (two paragraphs, risk-based):
9.1: "Where the secondary packaging is not opened, only the packaging material used needs to be retained, as there is no, or little, risk of product mix up."
9.2: "Where the secondary packaging is opened, for example, to replace the carton or patient information leaflet, then one retention sample, per packaging operation, containing the product should be taken, as there is a risk of product mix-up during the assembly process. It is important to be able to identify quickly who is responsible in the event of a mix-up... as it would affect the extent of any resulting recall."
The 2026 text (six clauses plus sub-clauses, prescriptive):
9.1: "Physical samples of packaging materials used in the re-packaging process (e.g. labels, carton, patient information leaflet, other package inserts) should be retained for the duration of the shelf-life of the re-packaged finished product."
9.2: "Reference samples of the re-packaged product are not required."
9.3: "A retention sample of the re-packaged finished product should be taken for each re-packaging operation and retained for at least one year after expiry date."
9.4: "The retention sample should represent the re-packaged finished product released for the market and should include the primary package as well as the secondary package. In cases where the secondary package is not opened, only the packaging material used needs to be retained."
9.5: "Where it is duly justified that a physical retention sample... cannot reasonably be retained, and this has been agreed in advance with the competent authority, a photographic/digital sample may be retained."
9.5.1–9.5.4: "the digital sample must be a "complete record," allow "full visual examination" and investigation "equivalent to that possible with a physical retention sample," comprise high-quality photos with traceability to the batch packaging record, capture all data on primary and secondary packaging (batch number, expiry), "show evidence that the safety features have been applied and allow the identification of information in braille," and, where stored electronically, "comply with the principles of Annex 11.""
What that shift actually means: five moves
1. From "assess the risk" to "here is the rule." The old text told you to think about mix-up risk and act proportionately. The new text simply tells you what to retain. The regulator has done the risk assessment for you and codified the outcome. Less interpretive latitude; less room to argue a lean local approach.
2. Packaging-material retention is now explicit (9.1). Retaining labels, cartons, PILs and inserts for the full shelf-life of the re-packaged product is now a stated Section 9 obligation, not something inferred from the general packaging-material clause (3.2). "We only keep the artwork we thought was risky" no longer holds.
3. Reference samples of the re-packaged product are explicitly not required (9.2). A genuine relief clause. If any local SOP currently mandates reference samples of re-packaged units, you can retire that requirement; the product was already manufactured and released, and the concern is verifying market presentation, not re-testing.
4. A retention sample for every re-packaging operation (9.3). This is the tightening. The old trigger was "secondary packaging opened." The new baseline is a retention sample per re-packaging operation, held one year past expiry, with 9.4 preserving the sensible carve-out that if the secondary package isn't opened, retaining the packaging material used is sufficient. Note the deliberate wording shift from "packaging operation" to "re-packaging operation": the sample is tied to the operation, not merely to the product or batch number. Same batch, different markets, different language versions, different runs means different operations, which means different samples.
5. The digital-sample option is entirely new (9.5). For the first time you can hold a photographic/digital retention sample instead of a physical one, but read the guardrails first. It is an exception, not a storage-saving default: it requires that a physical sample "cannot reasonably be retained," it must be duly justified, and it must be agreed in advance with the competent authority. Once you go digital, the image becomes a regulated GMP record and inherits full Annex 11 obligations: integrity, access control, backup, retrieval, and protection from uncontrolled change over the whole retention period.
The housekeeping changes (don't skip these: they touch your document references)
These are non-operational, but they will show up in an inspector's cross-check of your SOP citations:
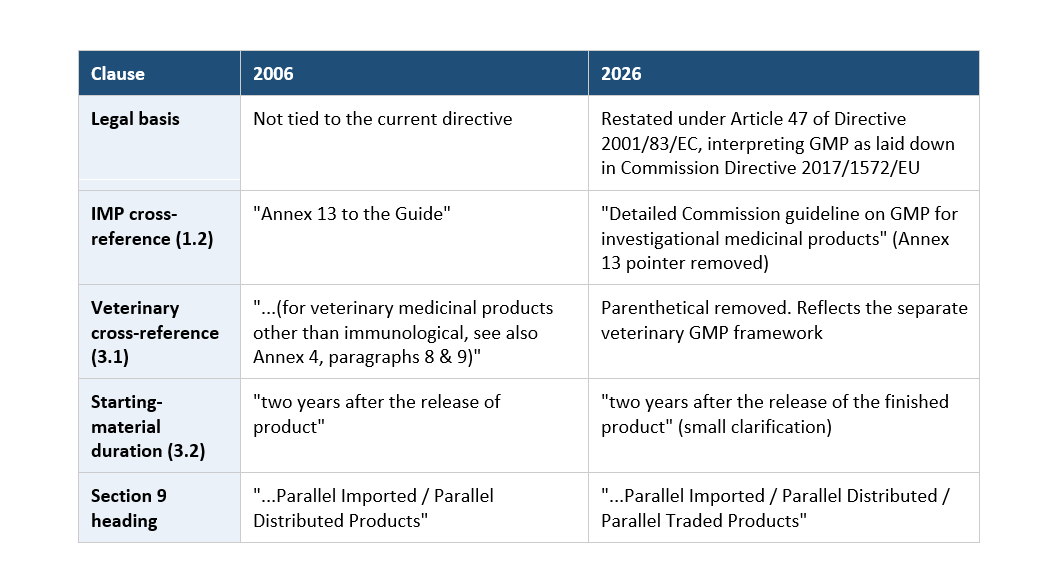
The scope pushback worth flagging: the Section 9 heading and the "reasons for changes" now name parallel traded products explicitly, but clause 1.3 still reads "parallel imported/distributed." Don't let the residual 1.3 wording narrow your scope. If you handle parallel trade through any commercial or regulatory route, treat it as in scope.
Why Now? The Context and Purpose Behind the Change
It helps to understand why the Commission reopened a text that had sat untouched since 2006. It tells you how to read the intent, and where inspectors' attention will land.
Trigger: the human/veterinary GMP split.
The immediate driver was legal housekeeping. Veterinary GMP has been carved out into its own framework: Commission Implementing Regulations (EU) 2025/2091 (veterinary GMP) and (EU) 2025/2154 (active substances used as starting materials for veterinary products), both adopted 17 October 2025 and applicable from 16 July 2026. Once veterinary medicines have their own legal base, the human GMP guide can no longer carry veterinary cross-references. That is exactly why the Annex 4 veterinary parenthetical vanished from clause 3.1, and why the legal basis is restated against Directive 2017/1572/EU. At the EMA's update to the 2026 ISPE European Annual Conference in Copenhagen, the Annex 19 revision was described precisely this way: an update following the separation of veterinary GMP requirements in the new implementing regulations.
Opportunity: modernise a 20-year-old section.
The Commission used that mandatory reopening to fix a section that had aged badly. The 2006 Section 9 was written for a simpler parallel-import model and leaned on the reader to judge mix-up risk. Twenty years of parallel-trade practice (multiple markets, multiple language versions, third-party re-packagers, and the Falsified Medicines Directive's safety features and braille requirements layered on top) made a risk-narrative clause inadequate. The rewrite converts judgement into defined, auditable obligations and, for the first time, tells re-packagers exactly what a retention sample must contain: primary and secondary packaging, safety-feature evidence, braille legibility.
Direction of travel: this is one tile in a much bigger mosaic.
Annex 19 is a small, early move in a broad modernisation programme run by the EMA GMP/GDP Inspectors Working Group (GMDP IWG) under its 2026 to 2028 work plan. The heavy items (Chapter 1 Pharmaceutical Quality System, Chapter 4 Documentation, a substantially rewritten Annex 11 Computerised Systems, and a brand-new Annex 22 Artificial Intelligence) are working through consultation now, with most deadlines set around Q4 2028 and a comprehensive revision of the EU GMP Guide anticipated. Read in that light, the new digital/photographic sample option in 9.5 is not a one-off. It is the same digital-records philosophy driving the Annex 11 rewrite, which is why 9.5.4 hooks straight back into Annex 11. The regulator is consistently signalling that electronic evidence is acceptable, provided the data-integrity scaffolding is genuinely there.
Not new in practice, but the door may now be wider.
One point worth saying plainly: in our knowledge and experience, it is sometimes possible, for small-batch, high-value products, to agree an exemption from holding the physical retention sample, relying instead on the reference sample alone plus a defined evidence package: detailed photography, dimensional testing, and artwork overlays, agreed and approved with the competent authority and documented in the Marketing Authorisation. It has never been a right and it has never been easy, but it has been done. What the new 9.5 does is potentially open the door to such discussions more readily, by putting a codified pathway on the page instead of leaving it entirely to bespoke negotiation.
Why pursue it at all? Simple commercial sense. For the right product, a retained unit is a unit not released to a patient and not sold, and at these values that cost recurs with every operation. But the door only opens to a very strong justification, and in practice that means a genuinely compelling combination of factors: prolonged synthesis and long manufacturing lead times, high manufacturing cost, low yield, small patient populations, and a relatively short shelf life. Absent that kind of profile, expect the answer to be no, and expect the evidence package to carry the entire weight of the argument. Something to think about, not something to assume.
The practical takeaway from the "why": treat this as a precedent, not an isolated tweak. The way you justify and control a digital retention sample now is a dry run for how you will handle the far larger digital-evidence expectations coming through Annex 11.
Weighing up a digital-sample case, or sitting on a legacy high-value-product exemption? A gap assessment will tell you exactly which SOPs and processes need updating, and whether your existing arrangements still hold up against the new framework.
Don't Forget: This Reaches Into the UK Too
Easy to file this under "EU problem." That would be a mistake. Post-Brexit, EU GMP guidance does not automatically become UK law. The UK's binding requirements flow from the Human Medicines Regulations 2012 and the MHRA's Rules and Guidance for Pharmaceutical Manufacturers and Distributors (the Orange Guide), and the MHRA can diverge where it chooses. But there are four reasons a UK operator should treat the new Annex 19 as live:
1. Any EU-facing product is squarely in scope. Great Britain is now a third country to the EU. If a UK site manufactures, imports, re-packages or arranges QP certification for product placed on the EU/EEA market, that product must meet EU GMP, including the revised Annex 19 from 24 September 2026, irrespective of what the Orange Guide says. For parallel trade specifically, GB to EU flows are exactly the operations Section 9 now governs.
2. Northern Ireland and the Windsor Framework. The MHRA remains the competent authority across the UK, and under the Windsor Framework (from 1 January 2025) all UK medicines, including NI, are MHRA-authorised, carry "UK Only" labelling, and sit outside the EU Falsified Medicines Directive for UK supply. But an NI-based QP named on a Northern Ireland GMP authorisation may still certify batches for both the UK and the EU markets. Where an NI operation certifies or re-packages for the EU market, EU Annex 19 applies to that product. The dual-market NI position is precisely where this gets missed.
3. Plan on the MHRA following the EU/PIC/S line. Our view: build the new Section 9 logic into your quality system now, even for GB-only operations. Treat UK alignment as the expected direction, not a possibility to wait on. The UK is a PIC/S member and the Orange Guide has historically reproduced EU GMP almost verbatim to preserve a shared quality language for trade and mutual recognition, so guidance drifts toward this text rather than away from it. There is no automatic adoption mechanism and the exact timing sits with the MHRA, but that is a reason to move first, not a reason to defer.
4. UK parallel import is the same problem in a different jacket. GB runs its own parallel-import regime (Parallel Import Licences / PLPI), and MHRA GMP expectations for re-packaging retention samples mirror the EU model. The operational lessons of the new Section 9 (a retention sample per re-packaging operation, packaging materials kept for shelf-life, samples that capture primary and secondary packaging with safety-feature and braille evidence) are simply good parallel-trade practice, and read across to UK domestic operations regardless of which guide is technically binding.
Bottom line for UK sites: don't wait for the MHRA to reprint it. If you touch EU-bound or NI dual-market parallel trade, you are already inside the EU version's scope. If you only touch GB, adopt the logic anyway as forward alignment and good practice.
Part 2: Ten Questions for Your Impact Assessment
1. Do we perform, or contract out, any re-packaging of parallel-imported, parallel-distributed or parallel-traded product, and for which market? Map each flow to its destination: EU/EEA-bound and NI dual-market product is already inside the revised EU Annex 19; GB-only product should adopt the same logic as forward alignment. (If you touch none of it, document that conclusion and stop; the rest is housekeeping.)
2. Does our current retention-sample SOP trigger on "secondary packaging opened," and if so, does it now under-comply with the "each re-packaging operation" baseline in 9.3?
3. How do we define a "re-packaging operation" in our records, and can we demonstrate a distinct retention sample (or justified packaging-material retention) for each one, across markets, language versions and separate runs?
4. Do we retain all re-packaging materials (labels, cartons, PILs, inserts) for the full shelf-life of the re-packaged product, or only a risk-selected subset? (9.1 is now prescriptive.)
5. Does any SOP still require reference samples of re-packaged product that we can now retire under 9.2, and is that removal itself justified and documented?
6. Does our retention sample composition capture primary and secondary packaging, plus safety features and braille evidence, sufficient for a future labelling, mix-up or falsified-medicines investigation?
7. Are we considering the photographic/digital sample option, and if so, can we actually evidence that a physical sample "cannot reasonably be retained"? (It is an exception, not a convenience.)
8. If we go digital, are we ready to obtain prior competent-authority agreement, and is our image store Annex 11-compliant: integrity, access control, audit trail, backup, retrieval over the full retention period?
9. Do our quality/technical agreements between MAH, re-packager, importer, batch-release site and storage site correctly allocate the new per-operation sampling, packaging-material retention and (if used) digital-sample responsibilities, and QP access to them?
10. Do all our SOPs, forms and templates cite the correct legal basis (C(2026) 4135 / Directive 2017/1572/EU) and drop the obsolete Annex 13 and Annex 4 cross-references?
Answering these ten questions properly is exactly what a gap assessment is for. If you'd rather have someone map your SOPs and processes against the new Section 9 than start from a blank page, that's what we do.
Part 3: What to Look At for Change Control
Run this as a planned change, opened now and closed inside the three-month window (before 24 September 2026). The scope of your change record should reach:
● Gap assessment against the new Section 9. The impact-assessment answers above become the objective evidence of your gap analysis.
● Regulatory / classification impact. Confirm this is a guidance revision requiring procedural alignment, and determine whether it is a "like-for-like" documentation update or a genuine process change for your parallel-trade operations (for most re-packagers, the latter).
● Documents in scope. Reference and retention SOP; re-packaging SOP(s); packaging-material retention procedure; batch packaging record requirements; sample indexing / traceability records; and any local forms specifying reference samples of re-packaged product.
● Cross-reference sweep. Every SOP that cites Annex 19, Annex 13 or Annex 4 needs a citation check, not just the retention SOP.
● Quality/technical agreements. Flag each agreement in the parallel-trade chain for review and, where needed, controlled amendment (this often has the longest lead time, so start early).
● Digital-sample decision gate. If in scope, a sub-record covering justification, the competent-authority engagement, acceptance criteria (photo quality, angles, data captured), Annex 11 system qualification/validation, and the file/archive strategy set out below. This is the part most people underestimate.
● Training and effectiveness. Who needs retraining (re-packaging operators, QA sample custodians, QP), and how effectiveness will be verified.
● Implementation deadline and owner. A defined target completion date inside the window, with a named owner, so the change is demonstrably controlled rather than reactive.
An inspector's fair question after 24 September is simply: "Show me the change record." The goal is that it already exists, and is closed.
If you are permitting digital samples: the questions that actually decide whether it works
A digital retention sample is only as good as its fidelity and its durability. 9.5.1 demands a record that supports investigation "equivalent to that possible with a physical retention sample," and 9.5.3 demands evidence that safety features have been applied and that braille can be identified. That is a high bar, and it is won or lost on decisions most SOPs never mention. Your change control should force answers to these before you go live:
● How will you deliver and retain it? Capture is the easy half. The hard half is holding a controlled, tamper-evident, retrievable record for the full retention period (shelf-life plus one year, and longer for some products). Who captures, who verifies against acceptance criteria, and where does it live?
● How durable is your file architecture? Plan for format obsolescence and media decay across a multi-year horizon: fixity/checksums to prove the file has not silently corrupted, a migration policy, backup and disaster recovery, and Annex 11 audit trail and access control that persist for the whole life of the record, not just the year you set it up.
● Which file formats, and why? This is a genuine trade-off you must justify, not default.
○ Raw / lossless (RAW, TIFF, DNG) for high-precision, forensic-grade detail. The safe choice where fine features must survive, at the cost of very large files and format-longevity management (proprietary RAW especially needs a migration plan).
○ Compressed JPEG to save space, but be honest about what compression destroys. A one-third-compressed JPEG at roughly 200 dpi may look fine on screen and be worthless for the job: lossy artefacts and low resolution are exactly what wipe out fine linework.
● Can you actually resolve the security detail? Put it to the test before you commit a format. Can you pick up the micro-printing? Can you resolve the anti-counterfeit / print security features, the guilloché, the microtext, the colour-shift or covert elements, at the resolution and compression you have chosen, under the lighting and angles you have specified? If the answer is "not reliably," your digital sample fails the 9.5.1 equivalence test and the 9.5.3 safety-feature test, and you do not have a compliant sample. You have a photo.
Worth saying plainly: a poorly specified digital-sample process is worse than keeping the physical sample, because it looks compliant until an investigation needs the one detail your compression threw away. Decide the format against the hardest feature you might ever need to examine, then validate it against a real re-packaged unit.
Part 4: Four Practical Options to Consider
Option 1: The "not in scope" close-out (lowest effort).
If you don't re-package parallel product, do the minimum properly: a short documented assessment concluding Section 9 doesn't apply, plus the citation/cross-reference housekeeping (C(2026) 4135, Directive 2017/1572/EU, remove Annex 13/Annex 4 pointers). One controlled document update, closed. Don't over-engineer it.
Option 2: Align procedures, stay fully physical (the default for most re-packagers).
Update the re-packaging and retention SOPs to the prescriptive Section 9 model: per-operation retention samples at one year post-expiry, full packaging-material retention for shelf-life, primary and secondary composition, and removal of any now-redundant reference-sample requirement for re-packaged product. No authority conversation needed; lowest regulatory risk; the trade-off is physical storage volume. This is the recommended baseline unless storage is a genuine, evidenced constraint.
Option 3: Adopt the digital-sample route for defined, justified cases.
Where physical retention is genuinely impractical (very high-value product, extreme storage volumes, bulky presentations), build a digital-sample process: a documented justification standard, an early competent-authority engagement, defined photo-capture criteria (data on primary and secondary packaging, safety-feature evidence, braille legibility), a resolved file-format and archive strategy (see the change-control section above), and an Annex 11-qualified image repository. Higher upfront build, real ongoing storage saving, but only if you can pass the "cannot reasonably be retained" test. Don't reach for this as a convenience; a weak justification is an inspection finding waiting to happen. If you already run a negotiated high-value-product exemption (physical retention waived in favour of the reference sample plus photography, dimensional testing and overlays, agreed with the authority and captured in the MA), this is your moment to review whether that MA-level arrangement should be re-expressed against the codified 9.5 framework, and whether your evidence package still meets the equivalence and safety-feature bar. And if you don't yet hold one but your product profile fits (prolonged synthesis, high manufacturing cost, low yield, small patient population, relatively short shelf life), the new text may make this the right moment to open the conversation.
Option 4: Hybrid, plus contractual push-down.
Physical retention as the default, digital reserved for a defined, pre-agreed subset, combined with a review of every quality/technical agreement so the sampling, retention and (where used) digital-sample obligations sit clearly with the party actually performing the re-packaging, and QP access is explicit. Best fit for multi-entity / multi-market operations where responsibilities are currently blurred across MAH, re-packager and storage site.
Bottom Line
The 2026 Annex 19 leaves the everyday retention regime alone and concentrates its force on Section 9. If you touch parallel trade, the direction is unmistakable: less interpretive latitude, more prescriptive retention, and a tightly-fenced new digital option that trades storage space for Annex 11 obligations. The safe move is a controlled change opened now, a clear scope decision (in or out), and, if you want the digital route, an early conversation with your competent authority rather than a September scramble.
Talk to Us About a Gap Assessment
Not sure whether Section 9 catches your operations? Want someone to map exactly which SOPs and processes need updating before the September deadline, rather than guessing?
A gap assessment against the new Annex 19 gives you a clear, documented picture of where you stand and a prioritised list of what to change. Whether you need a quick scope call, a full gap assessment, or support taking a digital-sample justification to your competent authority, we can help.
This is a Just Ask briefing for general guidance and does not constitute formal regulatory advice for a specific product, site or authorisation. For a site-specific gap assessment, a digital-sample file/archive strategy, or a competent-authority submission, talk to us.Primary and contextual sources: Commission Decision C(2026) 4135 (revised Annex 19, 2026); EudraLex Volume 4 Annex 19 (2006); Commission Implementing Regulations (EU) 2025/2091 and (EU) 2025/2154 (veterinary GMP, applicable 16 July 2026); EMA GMDP Inspectors Working Group 2026-2028 work plan and EMA regulatory update, 2026 ISPE European Annual Conference, Copenhagen; MHRA Windsor Framework guidance for human medicines (in force 1 January 2025); MHRA Rules and Guidance for Pharmaceutical Manufacturers and Distributors (Orange Guide) / Human Medicines Regulations 2012.